The Anhydrite Process (sometimes called the Müller-Kühne Process) was developed for production of sulfuric acid, but also produced Portland cement clinker as a by-product, and for fifty years during the 20th century it contributed significantly to British cement production capacity. In this process, anhydrite (calcium sulfate) replaces limestone in a cement rawmix, and under reducing conditions, sulfur dioxide is evolved instead of carbon dioxide. The sulfur dioxide is converted to sulfuric acid by the Contact Process using a vanadium pentoxide catalyst. As regards cement manufacture, the process was quite distinctive, with thermodynamics and emissions considerably different from those of traditional manufacturing methods, and the plants were unusually well-described.There were three Anhydrite Process plants in the UK between 1930 and 1976. This article brings the three plants together and discusses the process in general.
Sulfuric Acid Manufacture
Sulfuric acid is a bulk chemical that is strategically important in a wide variety of chemical processes. In industrial countries, its production statistics are often considered as an indicator of the general level of economic activity. Historically, sulfuric acid was first produced by calcining labile sulfates such as those of iron. Subsequently, methods developed for oxidising the more readily-available sulfur dioxide to sulfur trioxide, which is then reacted with water to give the acid. The use of nitrate salts to oxidise sulfur dioxide was described in the alchemical works of "Basil Valentine" (Note 1) in the 16th century. Production on an industrial scale began with the invention of the Lead Chamber Process by John Roebuck in Birmingham, England in 1746. In this, sulfur dioxide is oxidised by air, using nitrogen dioxide as an oxygen carrier. In 1831, Peregrine Phillips patented the Contact Process, in which sulfur dioxide is directly oxidised by air using, initially, a platinum catalyst. The Contact Process, being cheaper to operate, and making a stronger acid, gradually superseded the Chamber Process. Both processes required a supply of sulfur dioxide.
Sulfur dioxide was usually produced as a by-product of the calcination of sulfide ores, such as pyrite and the ores of copper, zinc, lead etc., and later from regeneration of gas works "spent oxide" resulting from de-sulfuration of coal gas. The burning of elemental sulfur - e.g. from Italy - was also used to a small extent, but during the 20th century, particularly in concert with the development of the petroleum industry in the USA, sulfur extracted from deep wells by the Frasch Process became a cheap source and became dominant. The world trade in sulfur, transported in bulk in liquid form, became the critical stage in sulfuric acid manufacture. However, wartime conditions had a damaging effect on this trade. While sulfuric acid was in high demand for explosives manufacture in war conditions, the trans-Atlantic and other trade in sulfur was disrupted by enemy action.
The squeeze on sulfur supplies was particularly severe for Germany during WWI, and intense research was put into alternative sources. Calcium sulfate in the form of anhydrite or gypsum is a geological resource, often of high purity, which is available throughout Europe in almost unlimited quantities. W. J. Müller and H. H. Kühne (Note 2) of Bayer (later I. G. Farben) studied the thermodynamics and kinetics of thermal decomposition of calcium sulfate and found that decomposition temperatures could be dramatically lowered by addition of silica or clay minerals to tie up the resulting lime, and by addition of coke to lower the oxidation state of the sulfur. The reaction is driven forward by the irreversible combination of calcium with shale minerals to produce Portland clinker. An experimental plant was set up at Leverkusen in 1916 in conditions of secrecy: the work was not published until 1926 - Müller, Zeitschrift für Angewandte Chemie, 1926, 39 p 169. Müller had returned to academia in 1926. The addition of argillaceous materials could be adjusted so that the process yielded a clinker of Portland composition as a by-product.
Application to Cement Manufacture
Portland cement clinker is traditionally made by calcining and sintering a finely-ground mixture of limestone and clay or shale. Many minerals and reactions are involved, but the most characteristic overall reaction forms alite:
3CaCO3 + SiO2 → Ca3OSiO4 + 3CO2
The Anhydrite Process replaces the limestone with anhydrite, and the corresponding reaction is:
3CaSO4 + SiO2 + 1.5C → Ca3OSiO4 + 3SO2 + 1.5CO2
An interesting consequence is that the raw-material-derived CO2 evolution is only one half that of the traditional process.
Anhydrite Process Kiln Chemistry
In the process as originally designed, a dry ground rawmix was made comprising anhydrite (or gypsum), shale and/or ash, sand and coke. This was fed to a simple rotary kiln and burned to produce clinker from the hot end and an exhaust gas containing sulfur dioxide.
Firstly, at 500-700°C, the available carbon from the coke reduces about a quarter of the calcium sulfate to calcium sulfide.
Equation 1:- CaSO4 + 2C → CaS + 2CO2
Next, the rest of the calcium sulfate reacts with the sulfide and shale silica, in the presence of low-temperature melts, to produce belite and calcium oxide.
Equation 2:- 3CaSO4 + CaS + 2SiO2 → 2Ca2SiO4 (belite) + 4SO2
Equation 3:- 3CaSO4 + CaS → 4CaO + 4SO2
Finally, in the burning zone, most of the free calcium oxide reacts with some of the belite to form alite
Equation 4:- Ca2SiO4 + CaO → Ca3OSiO4 (alite)
The clinker is rapidly cooled in the usual manner to preserve the reactive high-temperature minerals. The exhaust gases are de-dusted and cooled by water-washing, further cleaned with mist precipitators, then dried by scrubbing with sulfuric acid. The gas now contains only sulfur dioxide (about 9%), carbon dioxide, a little oxygen and nitrogen+inerts. Air is then admitted to bring the sulfur dioxide to around 6.5%, ensuring a two-fold excess of oxygen for the oxidation.
Equation 5:- 2SO2 + O2 → 2SO3
Finally sulfuric acid is formed by reaction with water.
Equation 6:- SO3 + H2O → H2SO4
A simplified stoichiometry implies that one mole of calcium sulfate gives rise to one of calcium oxide (56.08 g) and one of sulfuric acid (98.08 g), so 86.27 g of a clinker containing 65% CaO could be made. This implies that in an efficient ideal system, sulfuric acid yield (Note 3) should be about 1.14 times that of clinker. In practice, there are losses from both process streams, but more from the multi-stage acid plant, so that acid/clinker ratios actually obtained were in the range 1.00-1.05.
Process Operations
Raw Milling
The anhydrite was received in a crushed state. Run-of-mine anhydrite is typically sufficiently dry that grinding energy was sufficient to remove any moisture (Note 4). The other components (shale, sand, coke) had to be dried and rotary dryers were used for this. The components were metered from bins by weigh-feeders to a collecting belt which fed the raw mill. Raw milling was always done by standard multi-chamber ball mills. The ground product was conveyed to blending bins or silos, from which material was drawn to a kiln feed hopper.

Burning
Material entered the kiln by various methods. Both exit and mid-kiln gas temperatures were measured, to ensure that the burn-out of the coke was under control. The kiln (A) was fired by coal or oil by the usual methods. The gas flow through the kiln was maintained by the suction of the acid plant blowers and controlled by dampers.
Gas cleaning and cooling
Initial de-dusting was done by cyclones (B), although electrostatic precipitators were originally tried. The gas was then washed and cooled with counter-current water in an earthenware-packed tower (C). This removed about 90% of the remaining dust. Some sulfur dioxide was dissolved in the wash water and this was mostly recovered in a second counter-flow tower (D) swept with cold air, this being returned to the cooled gas stream. The air-flow rate was controlled to obtain the required sulfur dioxide : oxygen ratio in the mixed gas. This stream still contained some dust and water mist, and these were removed in wet "mist precipitators" (E). The gas then entered counter-flow drying towers (F) in which remaining moisture was scrubbed with sulfuric acid.
Oxidation of Sulfur Dioxide
The clean dry gases passed through blowers (G) into heat exchangers (H) in which hot product gases were cooled while the input gas was heated to 400-410°C. The gas then entered a catalyst-packed converter (I). The usual catalyst was doped vanadium pentoxide. The oxidation is exothermic, so in order to prevent temperature from running out of control, heat had to be exchanged with the cold input gas. Several cycles of heat exchange and conversion took place. The blowers were sized to provide ample draught for the kiln system and to deliver the 200 kPa required to push the gas through the multiple resistant catalyst beds.
Formation of Sulfuric Acid
The cooled converted gases were finally scrubbed in a counter-flow tower (J) fed with 98% sulphuric acid to produce an oleum, which could be taken as product or diluted (K) with more dilute acid from the drying towers, or with fresh water. The final products were hot and required water cooling to bring them to handleable temperature for storage.
Treatment of Tail Gas
The remaining gas comprised mainly nitrogen, some oxygen and carbon dioxide from kiln fuel combustion, plus a small amount of unconverted sulfur dioxide. This was scrubbed (L), typically with ammonia or alkali, the resulting sulfites being decomposed to sulfur dioxide which was returned to the gas drying tower inlet.
Kiln Process Problems
While the acid plant was fairly tolerant of process variations, a fundamental flaw in the process was the sensitivity of the kiln system and the variability of the clinker quality. The carbon content of the rawmix constituted a knife-edge control problem; in Equations 2 and 3, the calcium sulfate and calcium sulfide (produced by reduction) must be in exact 3:1 balance. With insufficient carbon present, calcium sulfate would remain in excess. It would survive in the burning zone and cause a melt-down, with damage to refractories and cooler. With too much carbon present, calcium sulfide would remain in excess, and would end up in the clinker, causing a variety of quality problems. In either case, insufficient calcium oxide would be made available, lowering the effective lime saturation of the clinker, and dramatically diminishing alite formation. The extent of this effect is examined in the Appendix.
The optimisation of the clinker-manufacturing part of the process was of low priority. The kilns used were necessarily dry process (Note 5), but by the standards of the cement industry, their fuel consumptions were high and their outputs very low. This is partly due to the much larger inherent endothermic heat of reaction: 3.7 MJ per kg of clinker compared with 1.7 MJ for a conventional mix. However, this was exacerbated by the use of coarse rawmix, made with the mistaken idea that this would reduce dust pickup. By raising burning zone temperatures, this increased gas velocities and viscosities, thus increasing the potential for dust pickup. All these kilns were limited by dust generation. Conceivably, a single stage preheater might have dramatically improved performance, but all the kilns were simple tubes with no internals. In addition to poor performance, the kilns were also subject to the inevitable reduced availability associated with a very complex plant with multiple sources of potential mechanical failure. The majority of short stops were associated with downstream problems in the acid plant, while the long-term effect of these led to long stops for major refractory failure. Subsequent expansion brought production to a peak in 1968, before a major fall in the price of elemental sulphur and the rise in energy prices wiped it out, the last production being in 1976.
Manufacturing History
International Development
The following is a chronological list of the Anhydrite Process plants commissioned. I am aware that there were other plants in Germany and Poland, but I have failed to track them down. Any additions - please contact me. The capacity given is the annual clinker capacity in tonnes. Sulfuric acid production was typically 3-4% greater.
| Date | Plant | Owner | kilns | capacity | ||
|---|---|---|---|---|---|---|
| 1916 | Leverkusen | NRW | Germany | Bayer | 2 | 50000 |
| 1930 | Billingham | Co. Durham | UK | ICI | 3 | 187000 |
| 1937 | Miramas | Bouches-du-Rhône | France | French Government | 1 | 24000 |
| 1938 | Wolfen | Saxony Anhallt | Germany | IG Farben | 4 | 210000 |
| 1951 | Wizow | Lower Silesia | Poland | Zakłady Chemiczne Wizów | 2 | 116000 |
| 1952 | Linz | Upper Austria | Austria | Österreichische Stickstoffwerke | 1 | 66000 |
| 1953 | Coswig | Saxony Anhallt | Germany | VEB | 4 | 230000 |
| 1955 | Whitehaven | Cumberland | UK | Marchon | 5 | 380000 |
| 1955 | Widnes | Lancashire | UK | USAC | 2 | 190000 |
| 1953 | Phalaborwa | Limpopo | South Africa | Foskor | ? | ? |
The plant at Leverkusen was essentially experimental. W. S. Müller and H. H. Kühne began academic work on the thermal decomposition of anhydrite as early as 1907, and Bayer commenced the pilot plant in 1913, finally making acid in 1916, around the time that Bayer joined the federation that became I.G. Farben. During war conditions the emphasis was on acid production at any price, and clinker was all discarded. It was not until the 1920s that clinker suitable for cement manufacture was made. The plant finally closed in 1931. In 1949, Kühne published a paper giving an interesting account of difficulties and misunderstandings encountered in developing the process. The plant at Billingham was mainly predicated in the availability of surplus anhydrite, and a certain amount of assistance in commissioning came from I.G. Farben. The plants at Miramas and Wolfen were mainly focused on explosives manufacture during re-armament. The plants begun during the 1950s were the result of a temporary scarcity of sulfur. All except Widnes used their acid mostly internally for phosphate production.
The developments in the 1950s in Britain were largely the result of government interventions. The US government, in the context of the Korean War, placed sulfur exports under limited allocation - a move which turned out to be entirely unnecessary, since sulfur production easily matched demand. The UK government, finding that the UK allocation was only two-thirds of likely requirement, placed sulfur and sulfuric acid under statutory control on 8/1/1951, To ameliorate the situation, the government set up a £25m fund to invest in new plants using indigenous raw materials. This immediately stimulated interest in the otherwise costly Anhydrite Process. The Billingham and Widnes projects began as early as May 1951, the Whitehaven project even earlier. In the 1950s, the UK government continued to maintain pressure, to the point of coercion, for industry to use indigenous raw materials.
The fairly concerted demise of the process in the early 1970s was primarily the result of reduction in the relative cost of elemental sulfur. In the USA, increasing availability of ore-burning sulfur reduced the price of Frasch sulfur, and increase of desulfuration sulfur from gas and oil processes - notably at Lacq in France - further reduced the price of the element. The process, like conventional cement manufacture, was fuel-intensive, so the 1970s volatility in fuel prices exacerbated the situation. Anhydrite process plants were either converted to sulfur-burning, or, in the case of Widnes, shut altogether. The reduction in cement capacity occurred at the time of the energy crisis turn-down, and so had no effect. ICI produced an amusing film extolling the environmental benefits of shutting down its cement plant! (Note 6)
There is little immediate prospect of a revival, although it is conceivable that an anhydrite process with greatly enhanced efficiency and perhaps a sulfoaluminate cement product might be considered in the future. The process will also be increasingly attractive by virtue of the fact that its CO2 emissions are lower than those of the conventional Portland cement manufacturing process.
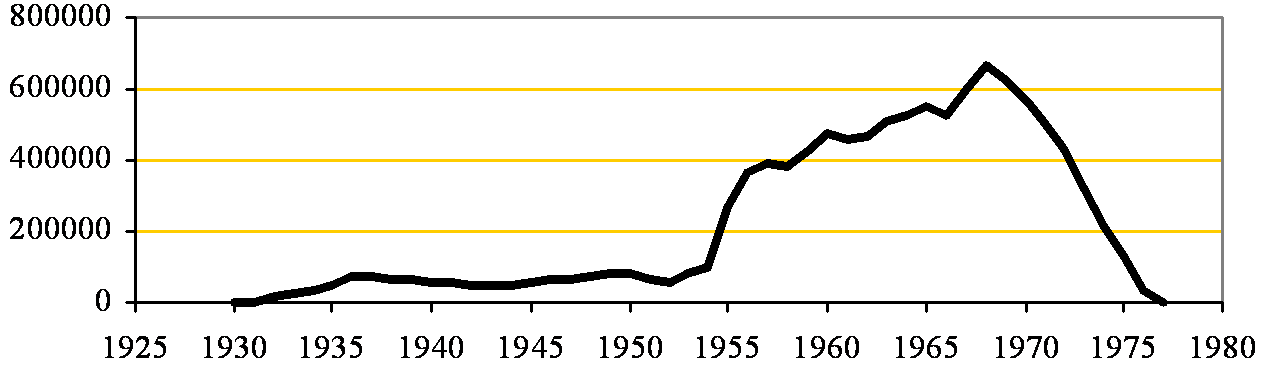
Billingham
The first British venture was at Billingham which was commissioned in 1930, using anhydrite mined 250 m below the plant. The plant was expanded with a second kiln in 1935 and a third in 1954, giving a total capacity of 187,000 tonnes a year. As mentioned on the Billingham page, the existence of an anhydrite resource below Haverton Hill was not a criterion (if known at all) for selecting the site for a chemical plant. Synthetic Ammonia and Nitrates (SA&N - a subsidiary of Brunner, Mond Ltd) proposed producing ammonium sulfate by BASF's Merseburg Process - reacting ammonium carbonate with calcium sulfate, and initially obtained anhydrite from Gotham in Nottinghamshire, and from Cumwhinton and Long Meg in Cumberland. In 1923, they became aware of the anhydrite seams at Hartlepool that had been proved by Charles Taylor Trechmann 20 years earlier. They paid the Warren plant to sink a mine on their site, and this supplied Billingham with anhydrite between March 1925 and June 1930. Meanwhile, boreholes were drilled during 1924 proving the Billingham deposit. The sinking of the mine began in September 1926, and production began in November 1927. This arrangement allowed Warren to take the brunt of the early technical difficulties of mining.
The mine accessed the main 5-6 m thick anhydrite seam 240 m below ground level. There were two 4 m diameter shafts: the downcast shaft conveyed two 4.5 t skips, while the upcast had double-decked cages for personnel and skips. All underground equipment, including large vehicles and the 27 m high primary crusher station had to taken down in small pieces and re-assembled at the bottom. The mine strata had the advantage of a fairly consistent 1:20 southeasterly dip. After secondary crushing on the surface, the anhydrite was fed to the acid plant by 190 m of belt conveyors and to the ammonium sulfate plants by 485 m ropeway.
The large majority of the material from the anhydrite mine went to the ammonium sulfate plant. Brunner, Mond & Co was one of the four constituent companies of ICI, formed on 18 December 1926, and a major expansion programme began in 1927. A small part of this was the construction of the sulfuric acid/cement plant, immediately east of the mine pithead. This was complete by the end of 1929, with one kiln, two parallel gas purification systems and four parallel converter/absorption systems. The acid plant was started up on sulfur burners on Saturday 14th December, 1930. The kiln was lit on Monday 10th February, 1931, and ran fitfully (less than 40 days' run) for the rest of the year. The total production for 1931 was: acid 10754 tonnes (mainly from sulfur burners), reject clinker 3343 tonnes, useable clinker zero. The average burning zone refractory life during 1931 was 11 days. The poor performance was put down insufficient control of feed chemistry and of kiln conditions, and poor mechanical condition of the kiln. The kiln did not run during the first 4 months of 1932, during which a major refit took place, and a team from I.G. Farben provided consultancy. The kiln was finally lit in mid-May under German control, and produced the first useable clinker. The kiln was stopped again in June to implement further major changes in the system, and finally re-lit in December. After this, the kiln ran with steadily improving performance, and the second kiln was installed during 1934, lighting up on Saturday 12th January 1935. During 1933 it had been established that the kiln was undersized for the acid plant, so little further modification of the acid plant accompanied the addition of the second kiln. The most obvious change was the replacement of the dry gas precipitators with cyclones. Experience with precipitators was slight in the 1920s, and clearly their use on gases with low moisture content and temperature over 600°C was hopeless; the 50% or more of the dust that passed them clogged up the downstream wet scrubbers.
The I.G. Farben recommendations to the plant in 1932 (Note 7) emphasised the importance of:
- the correct appearance of the kiln atmosphere.
- closeness of control of coal rate and kiln draught.
- closeness of control of %O2 in kiln atmosphere (1% recommended).
- constancy of composition of raw meal.
The "correct appearance of the kiln atmosphere" was a tenuous attempt to control the fuel/air ratio, as was the control of %O2. The knowledge of kiln gas composition at this stage consisted of Orsat-type analysis, which was highly retrospective and not suitable for minute-to-minute control. Actual control of coal and draught relied upon physical adjustments that corresponded only roughly to the actual mass-flows. For keeping the raw meal composition constant, only classical gravimetric analysis was available, with a long lead time, so again, short-term control was impossible. Bearing these impediments in mind, it should be no surprise that the plant had a faltering start. Having said that, the plant went on to pioneer more rapid means of data gathering. The first direct-reading oxygen meters were installed in 1949, using a thermal conductivity method, replaced in 1953 with paramagnetic meters. The plant also adopted XRF chemical analysis in the early 1960s.
Moses gives data on production up to 1950. This shows the improvement in performance over the period.
| Date | good clinker | reject clinker | acid | ||
|---|---|---|---|---|---|
| kiln 1 | kiln 2 | kiln 1 | kiln 2 | ||
| 1929 | 0 | 0 | 0 | 0 | 543 |
| 1930 | 0 | 0 | 3343 | 0 | 10754 |
| 1931 | 3444 | 0 | 960 | 0 | 10196 |
| 1932 | 15409 | 0 | 16897 | 0 | 32101 |
| 1933 | 42215 | 0 | 4484 | 0 | 44493 |
| 1934 | 39737 | 0 | 2946 | 0 | 41181 |
| 1935 | 14149 | 37084 | 1893 | 4166 | 54952 |
| 1936 | 31695 | 34108 | 3522 | 2177 | 65985 |
| 1937 | 22566 | 35541 | 2789 | 3091 | 60162 |
| 1938 | 34544 | 30755 | 1514 | 1115 | 65033 |
| 1939 | 29720 | 38419 | 1730 | 1352 | 70096 |
| 1940 | 41096 | 40268 | 1491 | 1246 | 82434 |
| 1941 | 43752 | 38683 | 757 | 709 | 82943 |
| 1942 | 43213 | 41141 | 658 | 290 | 86270 |
| 1943 | 39572 | 39665 | 279 | 401 | 79837 |
| 1944 | 45057 | 44264 | 272 | 133 | 88816 |
| 1945 | 45541 | 46707 | 321 | 472 | 90753 |
| 1946 | 52318 | 51535 | 263 | 259 | 102879 |
| 1947 | 48974 | 38609 | 495 | 272 | 86014 |
| 1948 | 49971 | 54197 | 1334 | 1562 | 103927 |
| 1949 | 54082 | 48369 | 1788 | 2016 | 100543 |
| 1950 | 54948 | 51620 | 2170 | 2320 | 102644 |
It's noticeable that, unlike most cement plants, the plant's production did not tail off towards the end of WWII. The lower amount of rejects during the war probably reflects a relaxation of quality standards. The operational problems of the process were largely ironed out during WWII, and the availability of government grants brought about the addition of kiln 3, in concert with the development of Widnes in 1953-1955, using a new rationalised acid plant.
The final closure of both the conventional and anhydrite plants in 1970 was part of a major structural change at Billingham that involved the use of elemental sulfur, the wholesale replacement of coal with gas as an energy source and raw feedstock, and the phasing out of ammonium sulfate as a fertiliser component, rendering the anhydrite mine redundant.
Widnes
The plant at Widnes was developed by the United Sulphuric Acid Corporation. This was a consortium consisting of ICI and ten of its sulfuric acid customers. The plant was set up at the time of the sulfur shortage scare, and was designed in concert with the expansion at Billingham, using common equipment designs. The setting up of a separate plant, rather than a larger expansion at Billingham, was done because of the high transportation costs of sulfuric acid. ICI did in fact briefly consider a sulfuric acid pipeline from Billingham to Merseyside (200 km). The site was adopted mainly for transportation logistics to the dense concentration of chemical industry customers in the Merseyside area. The adoption of a consortium was an indication of ICI's reluctance to finance what they knew from bitter experience to be a risky venture, susceptible to unpredictable market fluctuations. The initial consortium, formed in May 1951, consisted of:
- The Alumina Co. Ltd
- Thomas Bolton & Sons Ltd
- British Celanese Ltd
- British Enka Ltd
- Clayton Aniline Co. Ltd
- Courtaulds Ltd
- James H. Dennis & Co. Ltd
- Fisons Ltd
- Imperial Chemical Industries Ltd
- McKechnie Bros Ltd
- Transparent Paper Co. Ltd
By 1970, the consortium had shrunk to:
- 40.7% Imperial Chemical Industries Ltd
- 24.7% Courtaulds Ltd
- 24.4% Fisons Ltd
- 6.1% BICC
- 4.1% McKechnie Chemicals Ltd
The location of the plant for the convenience of product transport meant that the plant, uniquely, was far from its raw material source, and incurred the cost penalty of transporting anhydrite (1.7 tonnes per tonne of acid or clinker) from Cumbria. The Long Meg mine was a drift mine on the edge of the Vale of Eden 10 km northeast of Penrith. The rail connection was direct and straightforward, but it was a 160 km trip. The consortium was not involved in cement production: clinker was conveyed to a store, from which the adjacent grinding plant, operated by Blue Circle, drew clinker as required. This gave Blue Circle the decision whether to grind the clinker alone or to import conventional clinker to blend with it.
The plant (unlike the other two) maintained its initial capacity throughout, with two kilns similar to the large kiln 3 at Billingham. At the start of the 1970s, the plant was facing the same economic calculations as Billingham, and since existing decentralised acid plants could easily be adapted for elemental sulfur, there was no need to keep it going. The Blue Circle grinding plant kept going on imported clinker for a short time, and as a cement depot for a little longer, but then shut in favour of better-placed facilities.
Whitehaven
Marchon moved their chemicals business from London to Whitehaven having been bombed out in 1940, the object being to be remote from enemy action. As at Billingham, the existence of an anhydrite deposit was unknown at the time of choosing the site. After the war, the remoteness of the site became a major disadvantage for a company despatching its products and bringing in all its basic raw materials, among which was sulfuric acid, used in great quantity for phosphate production. An application to set up a pyrite-burning acid plant was turned down by the Board of Trade, but the government grants offered in 1951 stimulated interest, the plant then under construction at Linz being identified as a possible model - Franz Schon was Austrian.
Because the local coal mines (Note 8) penetrated the overlying Permian strata, the presence of anhydrite at the site was well established, and was announced to the Board in an "oh, by the way" fashion in 1951. Planning began immediately, and "the first sod" was cut on 24 May 1952. Interestingly, ICI had during WWII performed a "careful examination" of the Cumberland deposits, and concluded that "it is unlikely that a deposit providing more than 30 years supply of anhydrite could be worked near the Cumberland coast without entering into steeply inclined anhydrite which would probably extend beneath the sea, and it is considered that this is of no interest to the sulphuric acid industry." (Note 9). As it turned out, by far the largest of the Anhydrite Process plants was built on this deposit. Marchon was a disruptive new entrant. Attempts to collaborate with ICI failed and Marchon instead obtained the services of Hans Kühne - one of the originators of the process - who was well-versed in I. G. Farben production methods. Starting with a two-kiln plant, it grew rapidly to five kilns. with new developments occurring almost up to the time of retrenchment.
The mine had the inclined strata that so terrified ICI, but the dip turned out to be only a variable 1:7 to 1:20 and this conferred the considerable advantage that the deposit could be accessed by drift instead of the deep shafts needed at Billingham. Two seams were worked; the upper 4.5 m thick, and the lower 6-9 m thick. The 2.4 m interburden was shale interspersed with gypsum seams, and was originally side-cast, but was later partially used to replace the surface-quarried shale mainly on account of its much lower (2% vs 15%) moisture content.
The original two Edgar Allen kilns, started in late 1955, were supplemented by Vickers Armstrong kilns: No. 3, 1962 and Nos. 4 & 5, 1967, the final total capacity being about 1200 clinker tonnes per day, considerably greater than at any of the other plants world-wide. However, contraction began in early 1973 (a flat-out year in the cement industry) with the replacement of kilns 1 & 2 by a sulfur burner. Kiln 3 was replaced with a burner in 1975, and kilns 4 & 5 - the last in the industry - were replaced with sulfur burners in May 1976. The four-burner plant had a considerably increased acid production of 1600 t/day, and dramatically lower manning levels and costs.
The plant ground its own clinker and sold cement through Blue Circle's marketing division to markets mainly in Cumbria, swollen at the time by developments at Sellafield. With the 1967 expansion, surplus clinker was transferred by Blue Circle to various locations, including Wishaw, Widnes, Dunbar and Magheramorne. The acquisition of Wishaw by Blue Circle in January 1967 seems to have been specifically for this purpose.

